

All aromatic compounds were not created equal. I know this phrase will sound really strange, but it is unfortunately true. Reactions that work perfectly for the humble benzene do not work for other aromatic heterocycles like pyridines. Pyridines are specially interesting because they are present in thousands of biologically active compounds, from nicotine to the latest approved drugs.
Nitration is one of those reactions that do not work on pyridines. If you want to introduce an amino group at, let’s say, the C2 position of a pyridine ring, you can resort to SNAr reactions (pyridines are naturally amenable to those as they are pi-deficient) or couplings. Alternatively, you can convert the pyridine into an N-oxide (a concept similar to a protection/deprotection) or maybe attempt the old Chichibabin reaction. In most cases, however, you need the presence of «something» already in that C2 position, likely a halogen or maybe an oxygen that can then be converted into another suitable group. The issue is, you cannot introduce a nitrogen there directly, you need a pre-existing functionality in place before you can move forward.
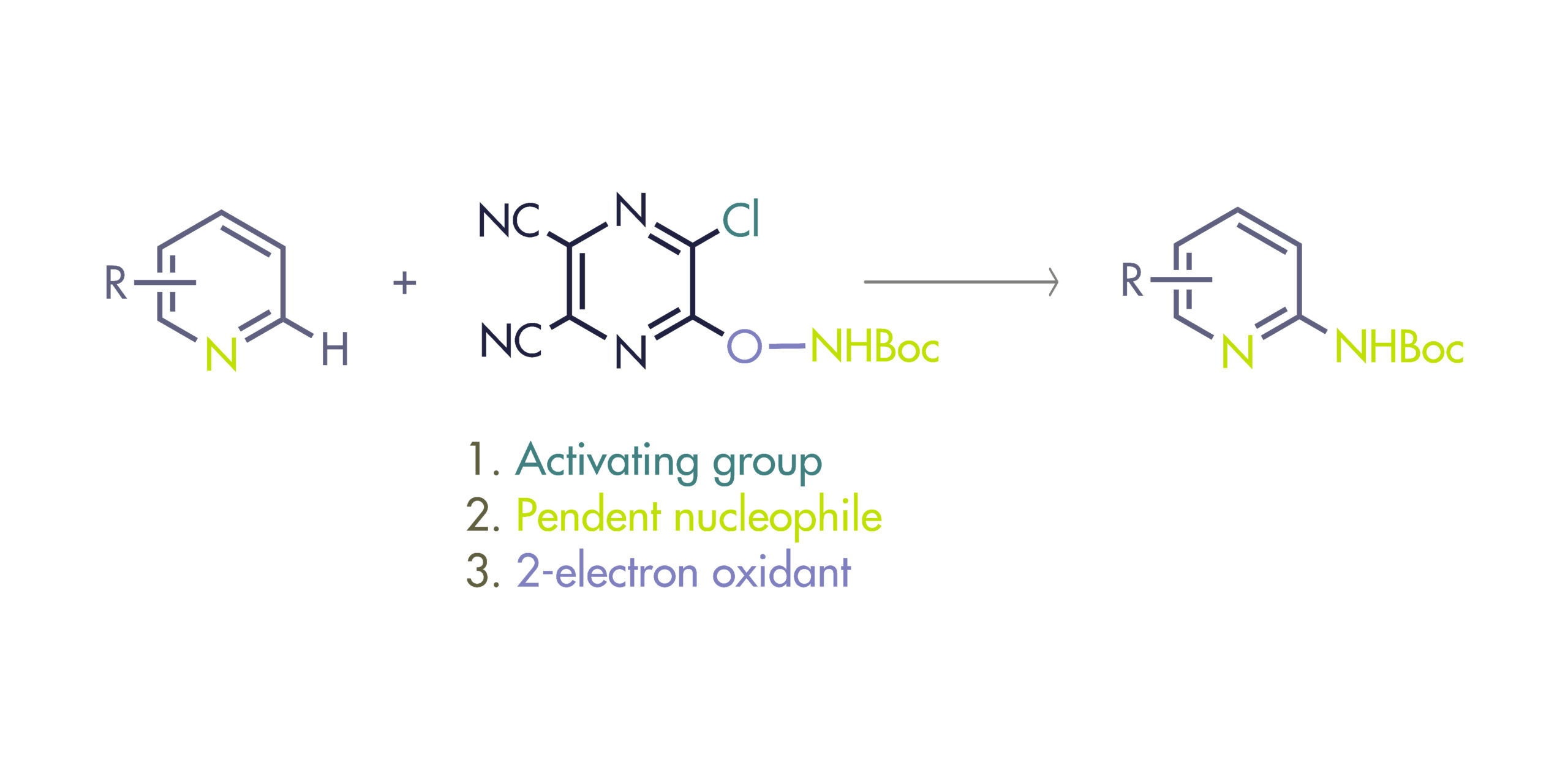
To solve this problem, Fier et al. (Merck, NJ, USA) have developed a reagent that selectively functionalizes the C2 position of pyridines introducing an amino group. The reagent is described as polyfunctional because it plays a triple role. First, it activates the nitrogen of the pyridine ring. Second, it transfers its pendent nucleophile to the C2 position of the pyridinium salt. And third, it serves as a 2-electron oxidant, so it can be removed safely and selectively to yield the final product.
The reagent can be prepared in hundred-gram batches by reaction of two cheap building blocks, 5,6-dichloropyrazine-2,3-dicarbonitrile and N-Boc hydroxylamine. It is crystalline, indefinitely stable in air, not shock-sensitive, and thermally stable in solid state up to 126 °C based on DSC analysis. The amination protocol is also simple: Mix the pyridine, the reagent (1.5 equiv) and N,O-bis-(trimethylsilyl)acetamide (BSA, 3 equiv) in dioxane at 80 °C for 3 h. Cool down to room temperature, remove the solvent and add Zn metal (5 equiv) in AcOH for 3 h. This second step promotes the mild cleavage of the reagent to reveal the 2-NHBoc pyridine product. The selection of the base was not trivial: typical inorganic or organic bases led to decomposition, but BSA worked by scavenging HCl through the generation of TMSCl.
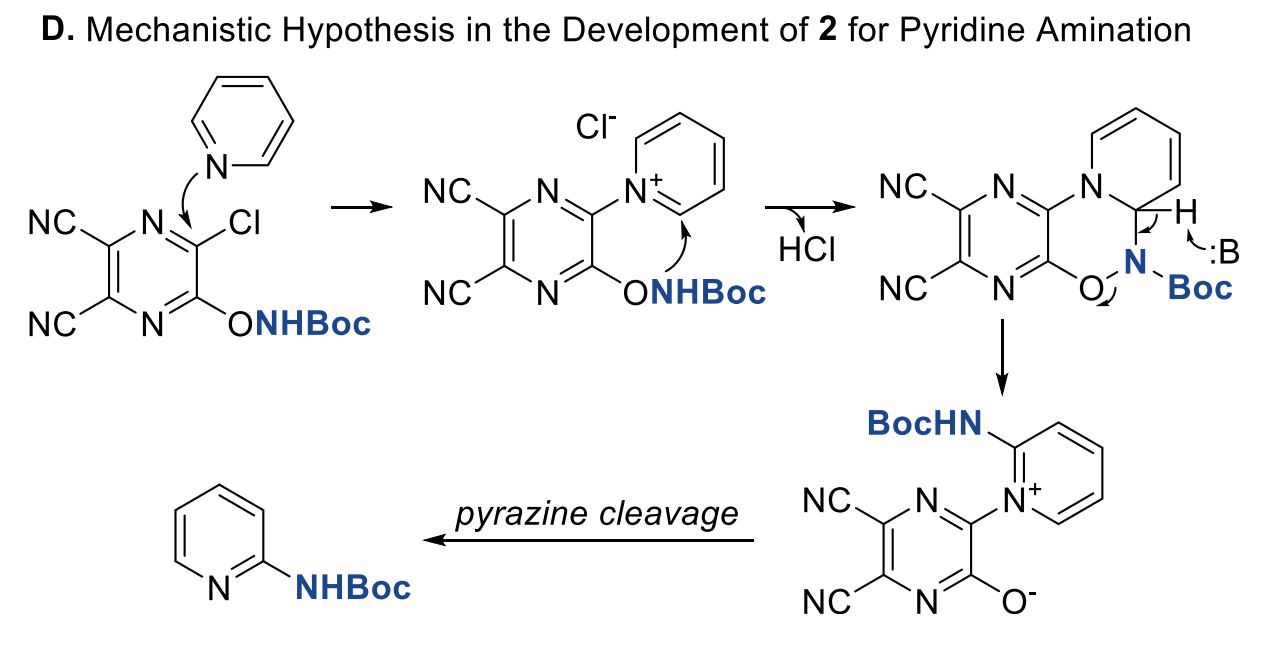
The protocol was tested on a number of pyridines with excellent results in most cases. Only with poorly reactive pyridines (for example, 4-substituted pyridines with electron withdrawing groups) the yields are low. As expected, the reaction can proceed through both the C2 and C6 positions and, when they are not equivalent, both regioisomers are observed. The Zn reduction step is problematic only on a single case, that is, when a dibenzylic ketone is present, but so far, no other problems have been found. The method can be even used as an LSF reaction, as demonstrated on Roflumilast as the substrate. In case you are interested, the authors have studied the mechanism in detail by NMR spectroscopy.
In the end, what do we have here? An interesting option to introduce an amino group in pyridines where the C2 position is not activated, an option to avoid regioselectivity problems in disubstituted pyridines, and a new reagent for LSF.
A Multifunctional Reagent Designed for the Site-Selective Amination of Pyridines J. Am. Chem. Soc. 2020, 142(19), 8614–8618. See: 10.1021/jacs.0c03537